





Abstract
3M syndrome is a rarely inherited autosomal recessive disorder caused by mutations in cullin-7 (CUL7), obscurin-like 1 (OBSL1), and coiled-coil domain containing protein 8 (CCDC8). It is associated with multiple dysmorphic features, including characteristic facial dysmorphism (a face that is triangular, full lips, frontal bossing, a nasal tip that is fleshy, long philtrum, protruding ears and macrocephaly), severe growth retardation prenatally and postnatally and normal intelligence. Although there are multiple skeletal manifestations of the syndrome, such as joint laxity, there is no mention of developmental dysplasia of the hip (DDH) to be associated with it anywhere in the literature. Therefore, we present the first case of bilateral DDH in a patient with 3M syndrome, which was managed similarly to other DDH cases with operative reduction, pelvic osteotomies, and femoral shortening, with a satisfactory outcome after 3 years of follow-up.
Background
3M syndrome is an autosomal recessive disorder that is rarely inherited (OMIM: 273750). It was described in 1975 and named after the three geneticists who first described it in 1975 [1]. Around 200 cases have been reported worldwide. 3M syndrome is caused by mutations in obscurin like cytoskeletal adaptor 1 (OBSL1), cullin-7 (CUL7), and CCDC8 [2]. OBSL1gene encodes a cytoskeletal adaptor protein, which is a member of the Unc-89/obscurin family. The protein contains multiple N- and C-terminal immunoglobulin (Ig)-like domains and a central fibronectin type 3 domain. Mutations in this gene cause 3M syndrome type 2. Alternatively, spliced transcript variants encoding different isoforms have been found in this gene [3]. OBSL1 clone is expressed highly in the ovaries and heart, moderately in the brain, testes and skeletal muscles, and lowly in other tissues tested. Biallelic variants in OBSL1 lead to 3-M syndrome 2 (MIM# 612921) and so far, up to 28 biallelic variants have been reported in the literature from different geographic populations [3]. Clinically, the 3M syndrome is characterized by characteristic facial dysmorphism (a face that is triangular, full lips, frontal bossing, a nasal tip that is fleshy, long philtrum, protruding ears, macrocephaly), severe growth retardation prenatally and postnatally and normal intelligence. The skeletal abnormalities are long tubular bones, thin ribs, short neck and thorax, pectus deformity, winged scapulae, fifth finger clinodactyly, reduced anteroposterior diameter of the vertebral bodies, hyperlordosis, joint laxity, and seldom delayed bone age [4]. Moreover, males sometimes have hypogonadism or hypospadias [5]. There is no mention of developmental dysplasia of the hip (DDH) to be associated with this syndrome. Therefore, we present the first case of bilateral DDH in a patient with 3M syndrome to ever be reported in the literature.
Case presentation
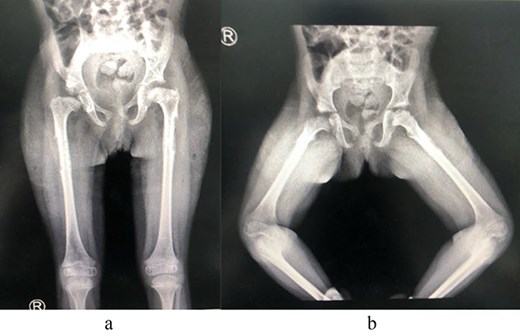
A 2-year-old girl was referred to our pediatric orthopedic clinic at XXX from a peripheral hospital for abnormal gait. On examination, the child had short stature and was thin. She had multiple dysmorphic features, which included a large head with frontal bossing and a triangularly shaped face, mid-face hypoplasia with depression in the nasal bridge and a fleshy nose tip, long philtrum, small pointy chin, micrognathia with large, prominent ears, and lordosis and waddling gait. A focused hip exam revealed bilateral limited hip abduction and leg length discrepancy. Images of the hips showed severe bilateral hip dysplasia (Fig. 1).
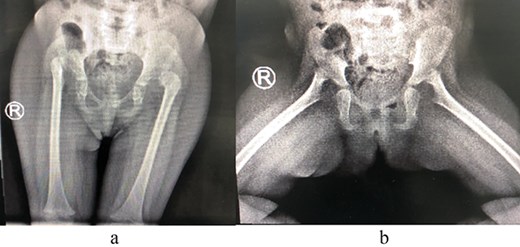
(a) Pre-operative AP view of the pelvis showing bilateral DDH. (b) Pre-operative frog-lateral view of the pelvis showing bilateral DDH.
A blood sample ethylenediaminetetraacetic acid (EDTA), was sent for whole exome sequencing (WES) for genetic diagnosis. It revealed a homozygous loss of function variant of OBSL1 (OBSL1:NM_001173408:exon3:c.1465C > T:p.R489X). According to American College of Medical Genetics and Genomics (ACMG) guidelines [6], it should be classified as a pathogenic variant. Homozygous loss of function (LOF) variants of OBSL1 is a common mechanism of 3-M syndrome 2, 612 921 [7].
The patient underwent left hip open reduction, pelvic osteotomy, femoral shortening and hip spica followed by open reduction, pelvic osteotomy, femoral shortening and hip spica for the contralateral side six months later (Figs 2–4), the intervention was similar to regular DDH cases, no differences were noted during the surgical management. The patient was followed up closely in the clinic, and she had the implants removed after one year (Fig. 5). The patient recovered fully without any apparent postoperative complications. She was followed up in the clinic for 3 years, and she recovered fully without any complications.
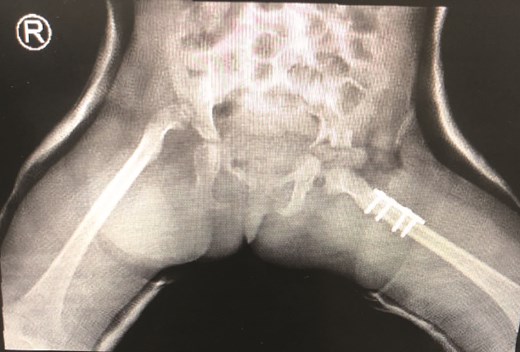
First stage correction of DDH.
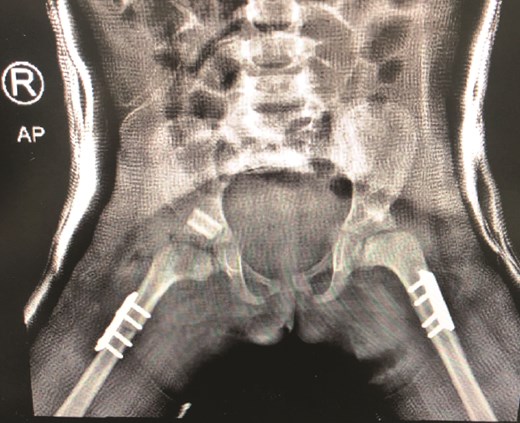
Second stage correction of DDH after 6 months from the first stage procedure.
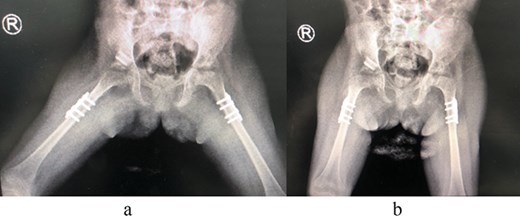
(a) Post spica application lateral view. (b) Post spica application AP view.
(a) Post removal of implant AP view. (b) Post removal of implant lateral view.
Discussion
The diagnosis of 3M syndrome was suspected based on clinical dysmorphic features and was confirmed with the genetic workup. In addition to the clinical features previously described, this is the first case to have bilateral DDH. To our knowledge, no other similar cases were reported in the literature, which might likely make it an incidental finding, however, it still raises the question of the possibility of an association of DDH to 3M syndrome; and, therefore, the need for careful examination for such a condition to avoid any delays in the diagnosis. Our report urges pediatric, genetic, and orthopedic physicians to always take into account the possibility of DDH and carefully examine the hips in syndromic patients in order to identify the presence of DDH and further study the possible association of DDH with 3M syndrome.
Conclusion
To our knowledge, this is the first case reported with DDH in a patient with 3M syndrome. We recommend screening 3M syndrome patients for DDH regularly in order to develop the most appropriate treatment plan.
Recommendation
We recommend that patients with 3M syndrome be carefully screened for DDH in order to avoid a delay in the diagnosis. We also recommend further research to understand the pathophysiology behind it and determine if there is indeed a relationship between the presence of this syndrome and DDH.
Author contributions
NA, MO, and IS contributed to the manuscript writing, JF revised the manuscript and wrote the case presentation.
Conflicts of interest
None declared.
Funding
This study did not receive any grants from funding agencies in the commercial, public, or not-for-profit sectors.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethical approval
This study was approved by the Ethics Committee of the XXX and can be provided upon request.
Consent to participate
Obtained for participation in the study/publication of data for research and educational purposes.
Consent to publish
Obtained for participation in the study/publication of data for research and educational purposes. Institutional approval was also obtained and is available upon request.
Use of AI
None declared.
References
Bioinformatics Software | QIAGEN Digital Insights. [

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}