





Abstract
Paragangliomas are rare neuroendocrine neoplasms arising from neural crest cells. Among these, primary gallbladder paragangliomas are extremely rare and are typically discovered incidentally during cholecystectomy. This case report describes a 37-year-old female presenting with biliary colic, who underwent laparoscopic cholecystectomy, which revealed a microscopic focus of paraganglioma. Immunohistochemical findings confirmed its benign, parasympathetic nature. Unlike sympathetic paragangliomas, these tumors are non-functioning and effectively managed with surgical resection. This case emphasizes the importance of considering gallbladder paragangliomas in differential diagnoses of biliary lesions and underscores the importance of further research into their pathogenesis, hereditary implications, and optimal management strategies.
Introduction
Paragangliomas are a distinct subtype of neuroendocrine neoplasms (NENs) known as neurogenic NENs, originating from the neural crest cells of the neuroectoderm. Collectively, they form the paraganglionic system, which includes the adrenal medulla and extra-adrenal neuroendocrine cell groups. Tumors located in the adrenal medulla are called pheochromocytomas, which often produce large amounts of catecholamines (CAs), leading to clinical symptoms such as high blood pressure and metabolic disturbances. Tumors outside the adrenal gland are called paragangliomas and can be further categorized based on their origin from either sympathetic or parasympathetic ganglia [1]. Paragangliomas originating from the glossopharyngeal and vagal nerves in the neck and skull base typically do not produce CAs due to their parasympathetic nature. In contrast, those arising from the sympathetic nervous system, which extends from the skull base to the pelvic floor, secrete CAs and are classified as sympathetic paragangliomas [2].
Sympathetic paragangliomas share characteristics with pheochromocytomas, including symptoms and histological features, such as the “pheochromogenic reaction.” Due to these similarities, their treatment approaches are essentially identical [2]. Paragangliomas can manifest in various locations throughout the body, affecting multiple organs in the head and neck, as well as the retroperitoneal and urinary systems [3]. Interestingly, paragangliomas originating in the gallbladder are extremely rare, with only a few reported cases. Most are incidentally discovered during cholecystectomy, and affected patients typically present with biliary colic. None of the reported cases showed signs of sympathetic system involvement or had a personal history of endocrine tumors or syndromes [4, 5].
Paragangliomas are exceptionally rare, with an estimated prevalence of 1 in 4500 individuals in the United States. These tumors most commonly affect individuals between the ages of 40 and 50 years old, with no significant sex predilection. However, parasympathetic paragangliomas are more frequently observed in women [6]. These tumors can develop sporadically or be part of a hereditary syndromes, with hereditary cases tending to present earlier in life and often exhibiting multicentric involvement. In children, sympathetic paragangliomas are uncommon, typically multifocal, and associated with hereditary syndromes.
Case report
A 37-year-old woman with a history of sleeve gastrectomy in 2014 presented to the clinic after visiting the emergency department with right upper quadrant (RUQ) abdominal pain. The pain had gradually worsened over three days, was non-radiating, and aggravated by fatty meals. It was also accompanied by nausea and vomiting. An abdominal ultrasound (US) revealed uncomplicated cholelithiasis, and the patient was advised to undergo a laparoscopic cholecystectomy.
The neoplastic cells displayed amphophilic cytoplasm with round nucelli and fine chromatin (Fig. 2).
On physical examination, the patient appeared well, alert, and oriented and was not in pain. She was neither pale nor jaundiced. Her abdomen was soft, lax, and not distended, with RUQ tenderness and a positive Murphy’s sign.
Her abdominal US revealed multiple gallbladder stones. No gallbladder distention, wall thickening, or peri-cholecystic fluid were observed. The common bile duct was within normal limits, measuring 0.2 cm.
Abdominal US conclusion: uncomplicated cholelithiasis
The patient proceeded with laparoscopic cholecystectomy, which was uneventful. No intraoperative gross abnormalities were noted.
Pathological findings
Gross examination of the gallbladder revealed an intact organ measuring 7.5 × 34 cm. The outer surface was gray-tan, smooth, and glistening. Upon sectioning, multiple black stones were identified, collectively measuring 0.8 × 0.6 × 0.4 cm. The mucosa was green-tan, partially velvety and partially sloughed, with a wall thickness of 0.2 cm.
Histopathological analysis revealed a single microscopic focus (<0.1 cm) within the adventitial fatty tissue, consisting of a well-circumscribed aggregate of tightly packed nested epithelioid cells (Fig. 1).
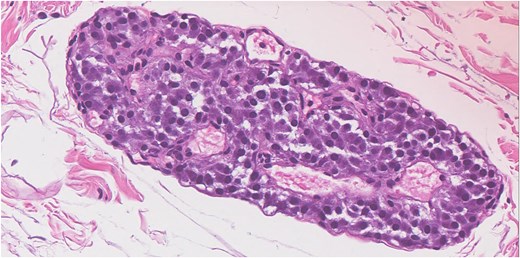
Hematoxylin and eosin stain show microscopic focus in the adventitial fatty tissue of a well-circumscribed aggregate of tightly packed nested epithelioid cells. The neoplastic cells have amphophilic cytoplasm with round nucelli and fine chromatin.
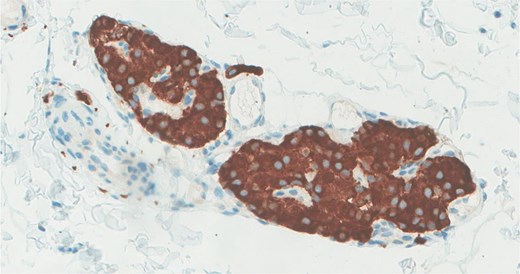
The neoplastic cells are positive for synaptophysin immunohistochemical stain.
Immunohistochemistry confirmed synaptophysin positivity, while S100 staining highlighted sustentacular cell (Fig. 3).
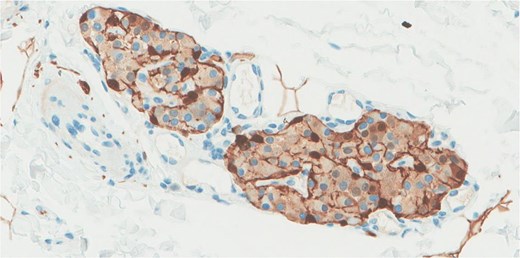
S100 immunohistochemical stain highlights the sustentacular cell.
The findings were consistent with paraganglioma. The cystic duct margin was negative, and additional sections revealed no further neoplastic foci.
Clinic follow-up
Two weeks after her surgery, the patient was seen in the clinic and reported doing well. She tolerated oral intake without nausea or vomiting and had non-active complaints. On examination, her abdomen was soft, lax, and not tender, and her surgical wounds were healing well. She was advised to undergo a contrast-enhanced CT scan of the abdomen and to follow-up as an outpatient.
Discussion
Primary gallbladder paragangliomas are exceptionally rare and not yet fully understood. These tumors likely originate from migrating paraganglia within the hepatic plexus, which innervates the gallbladder via the cystic plexus. The cystic plexus comprises both parasympathetic and sympathetic fibers. Based on current reports, primary gallbladder paragangliomas are considered non-functioning parasympathetic tumors, distinguishing them from pheochromocytomas and sympathetic paragangliomas, and are thus considered as benign. Therefore, incidental discovery during cholecystectomy is generally sufficient for treatment [7].
In contrast, sympathetic paragangliomas are more likely to be malignant and require staging before surgical resection, similar to pheochromocytomas. Although the gallbladder does receive sympathetic innervation, malignant paragangliomas originating from this organ have not been documented. Moreover, the limited number of benign paraganglioma cases lack long-term follow-up data. Given that these tumors typically present with biliary colic, they should be included in the differential diagnosis of gallbladder lesions. When multiple paragangliomas are identified, clinicians should consider the possibility of an underlying hereditary syndrome [8]. Despite the potential malignant transformation, further studies are required to determine whether genetic testing and surveillance for other endocrine tumors are warranted in these cases.
Conclusion
In summary, primary paragangliomas of the gallbladder are extremely rare, with limited clinical data available. Detecting these lesions requires a high degree of vigilance in preoperative imaging, as they exhibit significant and persistent enhancement on enhanced CT scans within the gallbladder. Diagnosis is critically dependent on histopathological analysis and immunohistochemical profile.
This report presents a case of primary gallbladder paraganglioma discovered incidentally following laparoscopic cholecystectomy for biliary colic. Given the limited published articles on similar cases, current evidence suggests that these tumors are parasympathetic, benign, and can be effectively treated with cholecystectomy. Based on neuroanatomy, we know that the gallbladder receives both sympathetic and parasympathetic innervation, but due to the extremely limited number of reported cases, the optimal follow-up remains unclear.
Further high quality clinical research is needed to establish standardized management guidelines and improve our understanding of these rare neoplasms.
Conflict of interest statement
None declared.
Funding
None declared.

{kind=link}
{kind=link}
{kind=link}