





Abstract
This retrospective case series evaluates treatment outcomes post-cochlear implantation in pediatric patients diagnosed with Cockayne syndrome (CS) and bilateral sensorineural hearing loss. Two female pediatric patients with CS type I underwent either bilateral or unilateral cochlear implantation. Visual reinforcement audiometry (VRA) and postoperative cochlear implant tolerance were the main outcome measures. Patient 1 demonstrated notable improvements in VRA results and school performance following bilateral implantation. Patient 2 experienced enhanced quality of life and environmental awareness post-unilateral implantation, despite a lack of objective VRA results due to developmental delay. The study underscores the benefits of cochlear implantation in CS patients, especially in patients who are post-lingual or with better cognitive function.
Introduction
Cockayne syndrome (CS) is a rare genetic condition with an incidence rate of <1 case per 250 000 live births in the USA [1]. It was first described in 1936 by Edward Cockayne in his work “Dwarfism with retinal atrophy and deafness”. It is caused by mutations in the ERCC8 (CSA) or ERCC6 (CSB) genes [2, 3]. These genes cause impairment in nervous system development characterized by a range of abnormalities such as photosensitivity, short stature, i.e. dwarfism, progeria-like appearance, learning delay, retinopathy, and progressive sensorineural hearing loss (HL) [4, 5].
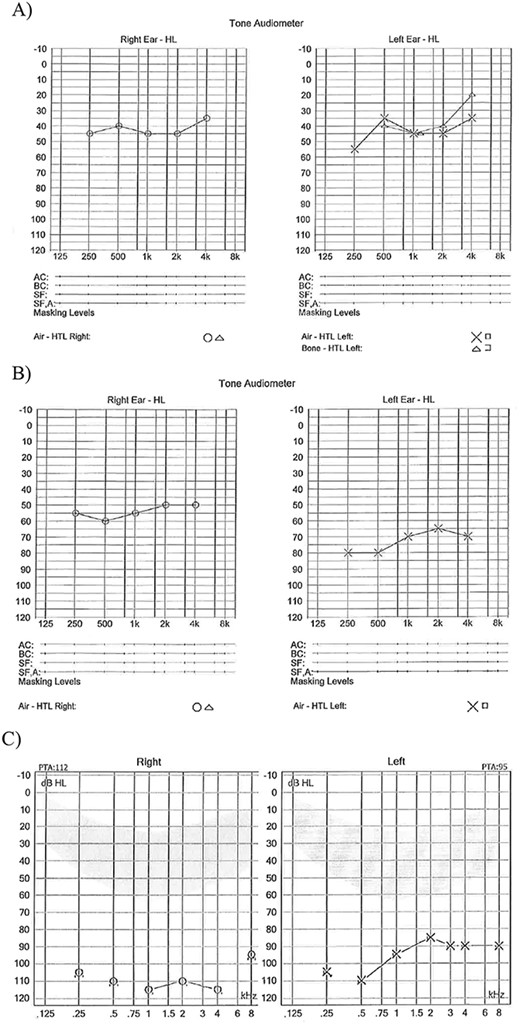
(A) Age 8 mild to moderate HL both ears; (B) Age 11 years moderate HL right ear and severe HL left ear; (C) Age 14 years profound HL both ears.
CS is incurable and there is no definitive treatment approach. The Cockayne Syndrome Natural History (CoSyNH) study was the first study designed in attempt to develop treatment guidelines. A total of 102 cases were prospectively reviewed and 44% (20 patients out of 45) had conductive or mixed HL. Of these, 21% (14/68) of cases presented neonatally and in 84% by 10 years of age [6].
HL is one of the most common manifestations of the CS occurring in more than half of the cases described in literature until this date [7]. Often audiometry in these patients is challenging due to their neurological and/or cognitive issues. These are thought to be the main reasons of poorly described HL in this cohort. In most of the cases patients present with bilateral HL, which tends to be sensorineural high-tone HL progressing to profound deafness over a course of years [5, 7].
Interestingly, early studies of temporal bone changes in CS have been shown to be consistent with age-related HL, such as loss of hair cells in pars superior, collapse of the endolymphatic duct of pars inferior, and corresponding changes in spiral ganglion [8]. This seems logical as CS is a disorder of premature aging. More recent histopathology findings show that most of the pronounced cochlear damage appears in the stria vascularis and spiral ligament instead of the spiral ganglion. These findings suggest that cochlear implantation could benefit patients with CS including those in the advanced stages of the condition with profound HL [9].
We describe case series of two pediatric patients with CS type I who had unilateral and bilateral cochlear implantation at the age of 5 and 14 respectively, and their treatment outcomes.
Case series
Case 1
Patient 1, a female with CS type 1, faced a range of challenges including learning difficulties, fatigue, gastrostomy feeding, and visual problems, in addition to her HL. Despite not having a diagnostic auditory brainstem response (ABR) at birth, she was presumably born with normal hearing. However, her family reported HL prior to her official diagnosis of moderate HL at age 10. Until the age of 14, she managed well with hearing aids (Fig. 1).
As her hearing deteriorated, she became heavily reliant on lip reading and struggled with basic tasks like using the phone. Her confidence at school declined, and she withdrew from social interactions. Her mobility also suffered, and she increasingly relied on a wheelchair.
Recognizing the impact of her HL on her quality of life, a hospital multidisciplinary team deemed her suitable for cochlear implantation as she met NICE criteria, and she underwent bilateral cochlear implantation at age of 14 with CP910s. Post-implantation, she experienced significant improvement, being able to communicate in full sentences and appreciate music. Her school performance improved, and she regained her confidence, even using the telephone comfortably.
A year after implantation, her speech testing using McCormick Toy Test showed remarkable progress, with a pass rate of 90% at 50 dB and nearly passing at 70% at 40 dB. Over the following 3 years, she continued to improve, regularly following up with the cochlear implant program. Her journey highlights the transformative impact of cochlear implantation in enhancing communication, social interaction, and overall quality of life for individuals with CS and HL.
She sadly passed away at the age of 19 due to multiorgan failure.
Case 2
Patient 2, a female diagnosed with CS type 1 at 1 year old, faced a multitude of challenges including profound bilateral sensorineural HL, congenital bilateral cataracts, developmental delay, and gastro-esophageal reflux. Despite these difficulties, she remained a cheerful child, living happily with her family. Her HL was detected early (failed newborn screening test), and she received binaural hearing aids at just 4 weeks old, although her benefit from them was limited.
An MRI head scan was performed at the age of 3 demonstrated significant progression with loss of cerebral, cerebellar, and brain stem volume (ex vacuo dilatation of ventricles, thin corpus callosum, and abnormal white matter signal).
By the age of 4, she could communicate with simple vocalizations but struggled with behavioral audiometry due to cognitive issues. ABR testing revealed profound HL. Despite her cognitive challenges, a multidisciplinary team deemed her suitable for cochlear implantation according to NICE Guidelines criteria, understanding that she might only appreciate environmental sounds with the implant.
She underwent successfully a left cochlear implantation Cochlear CI512 device at the age of 5. Intraoperatively her left cochlea was grossly abnormal (rotated). The decision to delay the right side implantation was made pending further imaging. Post-implantation, her family noticed positive changes as she responded to sounds, turned toward speech, and showed increased alertness and activity at school. While she did not proceed with a sequential cochlear implant, follow-up assessments using visual reinforcement audiometry showed no discomfort, and the implant functioned correctly according to neutral response telemetry.
Despite the limitations in her ability to link sounds with visual rewards, her improved response to auditory stimuli, direction awareness, and appreciation for music with the implant highlighted the benefits of cochlear implantation in enhancing her quality of life.
She sadly passed away at the age of 8 years due to multiorgan failure.
Discussion
CS presents unique challenges in hearing rehabilitation due to its rarity and complex clinical manifestations. Limited data on hearing outcomes in CS patients are available, primarily from small case series [5, 8]. In our study, both patients demonstrated positive outcomes post-cochlear implantation, aligning with existing literature.
The first patient, with progressive bilateral sensorineural HL, showed significant improvement post-implantation. Having been lingual prior to the procedure, she regained the ability to appreciate environmental sounds and speech. Similarly, the second patient, despite being pre-lingual and having severe developmental delay, benefited from cochlear implantation by developing environmental awareness.
These results are consistent with findings from previous studies, particularly in cases where patients were lingual prior to implantation. A paper describing pediatric patients with CS Type 1 and progressive HL showed favorable outcomes post-implantation, with improvements in both environmental sounds and speech perception. However, one patient experienced speech difficulties later, potentially due to natural progression of CS [5].
Similarly, studies involving adult CS patients who were lingual prior to implantation reported positive outcomes, with improvements in environmental sound perception. However, one patient faced challenges with speech perception, attributed to either auditory system deficits or cognitive impairment [10].
The variability in outcomes across studies may stem from differences in patients’ cognitive baselines and lingual status before implantation. Despite these variations, both of our patients and those in the literature experienced improvements in environmental awareness post-implantation, highlighting the subjective enhancement in their quality of life as reported by their families.
Overall, cochlear implantation offers tangible benefits for CS patients, improving environmental awareness and, in lingual patients, maintaining or enhancing speech perception. Collaborative efforts between healthcare professionals and families are essential in evaluating the potential benefits and outcomes of cochlear implantation in CS patients, especially those with cognitive challenges. Further research and larger studies are warranted to better understand the long-term efficacy and implications of cochlear implantation in this rare population.
Conclusion
Sensorineural HL management in patients with CS remains challenging due to complex of neurological and developmental issues. Patients with CS and sensorineural HL are more likely to benefit from cochlear implantation if they do not suffer from severe cognitive impairment and/or managed to develop speech prior to deterioration of their HL. It is crucial to work closely with the members of the multidisciplinary team and the family in the decision-making process to evaluate possible benefit from the cochlear implantation for patients with severe cognitive problems.
Funding
None declared.

{kind=link}