





Abstract
Inflammatory myofibroblastic tumor is an extremely rare neoplastic lesion with a predilection for aggressive local and recurrent behavior. The tumor tends to occur in the lungs of children and young adults, and although it can develop in older patients and other organs, this is extremely rare. Symptoms are nonspecific and depend on the location and size of the tumor. The gastrointestinal tract is rarely this mass’s primary site of origin, and the cecum is an even rarer location. We present the case of an otherwise healthy 55-year-old female who presented with an acute abdomen and a mass in her abdomen; after successful surgery, she fully recovered. Inflammatory myofibroblastic tumor causing acute abdomen was the final diagnosis.
Introduction
Inflammatory myofibroblastic tumors are rare lesions formed by the proliferation of fibroblastic–myofibroblastic cells [1]. They usually appear during infancy and youth with non-specific symptoms, and although they may show a benign appearance, they have an intermediate malignant potential [1, 2]. Therefore, surgery is the treatment of choice. These tumors are extremely rare in the colorectal segment, with fewer than 30 cases ever described in the literature [3].
We present the case of a 55-year-old female who presented with an acute abdomen and a mass in her abdomen; after successful surgery, she fully recovered. Inflammatory myofibroblastic tumor causing acute abdomen was the final diagnosis.
Case report
Patient is a 55-year-old female without any medical history; she suffered from sudden severe lower abdominal pain without nausea or vomits. As the pain persisted and did not improve, even with over-the-counter analgesics, she presented to the emergency room. We encountered a tachycardic, febrile (38 C) patient with lower abdominal pain with tenderness. A hard, palpable mass was detected in her lower abdomen, but no lymph nodes were found.
Complementary exams revealed normal leukocytes (9.86 × 109/l) without neutrophilia and an elevated C-reactive protein. Due to the pain and the palpable mass, a contrast-enhanced computed tomography (CT) was needed. It revealed a 15 × 15 cm heterogeneous mass that compromised the cecum and ileocecal valve; it also had mesentery involvement, with focal strands radiating into the mesenteric fat (Fig. 1).
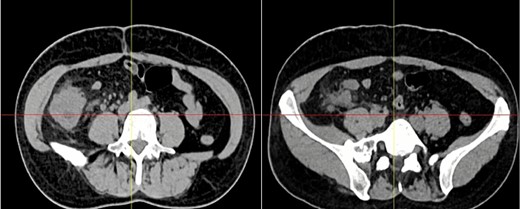
Abdominal CT, the mass is seen in the cecum and ileocecal valve.
Surgical consultation was needed with these findings, and after explaining it to our patient, surgery was planned.
At laparoscopy, multiple adhesions were discovered between the abdominal wall and omentum, which were released using an ultrasound energy device (Harmonic, Ethicon NJ). Then, the mass was unveiled; it measured 15 × 10 cm and compromised the mesentery, the cecum wall, and the ileocecal valve. The mesentery seemed hardened and had many lymph nodes. Due to this, a right hemicolectomy was performed; the ileum was stapled 10 cm away from the ileocecal valve using mechanical staplers (Echelon Flex, Ethicon NJ), and the colon was exposed and released. The ileocolic and right colic arteries were clamped as they branched from the superior mesenteric artery. Afterward, the hepatic flexure of the colon was released and stapled, and the mass was removed entirely through the umbilicus. After that, a side-to-side ileo-transverse anastomosis with staples was completed, and the enterotomy was closed with a 2-0 Polyglactin 910 Suture (Ethicon Coated Vicryl, Ethicon NJ). A drain was placed, and the procedure was completed without complications.
Pathology reported a 10 × 6 cm tumor. It expanded from the cecum into the ileocecal valve and had a soft exterior. Microscopically, it was formed by multiple myofibroblastic spindle cells with borderline malignancy. Numerous plasma cells and lymphocytes were also seen (Fig. 2). Immunohistochemistry was positive for actin and ALK (Fig. 3). Margins were free of tumors, and all lymph nodes were negative for invasion. Inflammatory myofibroblastic tumor was the final diagnosis.
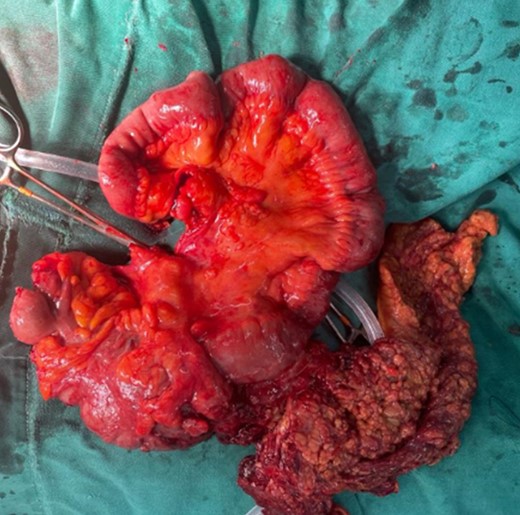
The mass is completely removed from the patient; it involves the cecum and ileocecal valve.
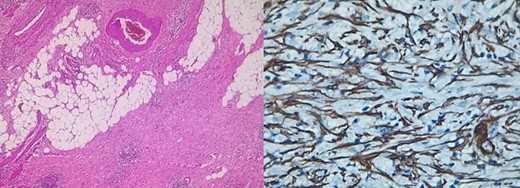
Immunohistochemistry: positive for actin and ALK.
The patient’s postoperative course was uneventful; she was on a liquid diet on postoperative Day 2, and after she passed gas and stool, she was discharged without complications. On follow-ups, she is doing well.
Discussion
Inflammatory myofibroblastic tumors are a rare subset of tumors first described by Harold Brunn et al. in 1939 [1]. They are formed by a combination of myofibroblasts or fibroblasts and a variable number of inflammatory cells (eosinophils, plasma cells, and lymphocytes) [1, 2]. They were once considered inflammatory benign tumors; however, they can show aggressive behavior with recurrences [2, 3]. Therefore, they are now classified as intermediate-grade neoplasms, which have a high recurrence rate after excision and exhibit low metastatic potential [2].
The etiology of this disease is still under study; however, they are believed to arise from chronic infections, autoimmune diseases (IgG4 disease), and trauma [1–3]. They typically affect children and young adults and have an indolent course [2]. They predominantly affect the lungs and abdomen (75%) [2, 3]. However, any body part, including the head, neck, chest, and central nervous sensations, can be affected [1, 3].
Symptoms are nonspecific and frequently associated with general inflammatory symptoms such as fever or malaise; nonetheless, they will depend on where the tumor develops and are associated with the tumor mass effect, swelling, and subsequent inflammation [2, 3]. In our case, the patient experienced acute abdomen and not obstruction due to the tumor, a rare event that has not been well described in the literature. For instance, tumors in the airways will cause dyspnea and epistaxis. In contrast, tumors in the intestine will cause obstruction [4].
Laboratory exams will be nonspecific as these lesions usually lead to an elevation of inflammatory parameters such as C-reactive protein and erythrocyte sedimentation rate, similar to other nonspecific surgical conditions [1, 5]. Nonetheless, imaging will help in the diagnosis and preoperative planning [1]. In our case, the patient suddenly presented with an acute abdomen and a palpable mass. After evaluation, C-reactive protein was elevated, and the CT revealed the tumor in the intestine.
CT or magnetic resonance imaging (will show the tumor as a heterogeneous or homogeneous mass depending on the organ in which it is found [1]. Therefore, they are often misdiagnosed as malignant neoplasms [1, 6]. Pathology is the only way to achieve a final diagnosis [1, 2].
The differential includes benign and malignant tumors such as gastrointestinal stromal tumor (GIST) (1 out of 100 000 adults per year), inflammatory fibroid polyp (0.1%–3.0% of all polyps in this body), lymphoma (0.2%–1% of all colonic malignancies), and adenocarcinomas (3.2 to 4.2 per 100 000 persons), among others [2, 4].
Pathology reveals a proliferation of fibroblasts and myofibroblasts, accompanied by mixed inflammatory infiltration by plasma cells, lymphocytes, eosinophils, and histiocytes [7]. Myxoid intercellular content, ganglion-like cells, necrosis, lymphovascular invasion, and high mitotic activity are considered adverse factors that worsen the prognosis [1, 7]. These tumors show positivity for smooth muscle actin and can be positive or negative for other myoids (desmin, transgelin, etc.) [1, 2, 8], as it was found in our patient.
Recurrence appears in up to 5% of all cases and is associated with abdominopelvic location, tumor size beyond 8 cm, and older patients [1, 2, 8]. Surgical resection with negative margins is the only treatment that can offer a complete cure; in cases where the tumor cannot be resected because of its location or because of the invasion of vital organs, antihumoral drug therapy such as tyrosine kinase inhibitors (crizotinib, etc.) can be used [1–3].
Conclusions
Inflammatory myofibroblastic tumors are rare mesenchymal tumors, and due to their rarity, their clinical features are still being studied. Rare presentations like our patient in which there is an acute abdomen with a mass should raise our awareness of this rare pathology and remind us that these infrequent pathologies should always be kept in the differential.
Conflict of interest statement
None declared.
Funding
None declared.

{kind=link}
{kind=link}
{kind=link}