





Abstract
Small cell carcinoma of ovary, hypercalcemic type (SCCOHT) is an unusual malignant tumor that most commonly affects young women. Unfortunately, it has a very poor prognosis. We describe here an unusual case of a Moroccan young woman with a left ovarian mass and a symptomatic hypercalcemia. Morphologically, there are some malignant tumors that resemble SCCOHT; thus, it is very challenging to diagnose, and immunohistochemistry has a great contribution in it. Hopefully, molecular tools and new therapies will improve the management of these cases in the near future.
Introduction
Small cell carcinoma of ovary, hypercalcemic type (SCCOHT) is a very aggressive malignancy with an early tendency to distant metastasis and a low 5-year survival rate. In general, the incidence of epithelial ovarian tumors is higher in older patients (>50 years), but SCCOHT is a rare tumor that tends to affect young women, with a mean age at diagnosis of 24 years. Herein we describe a case of this rare entity of a North African 22-year-old woman with a brief review of literature.
Case
A 22-year-old woman presented with chronic abdominal and pelvic pain associated with acute vomiting in the context of generalized asthenia.
An abdomino-pelvic ultrasound was performed showing a pelvic lesional process with a left-lateralized supra uterin location, with a mixed echo-structure, mainly of tissue, classified O-RADS 5 (Figs 1 and 2).
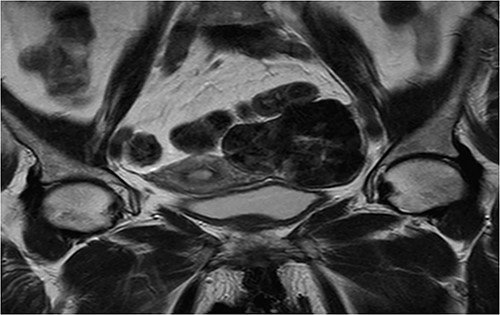
Coronal T2 sequence: left uterine mass, heterogeneous, in T2 hyposignal containing areas in hypersignal.
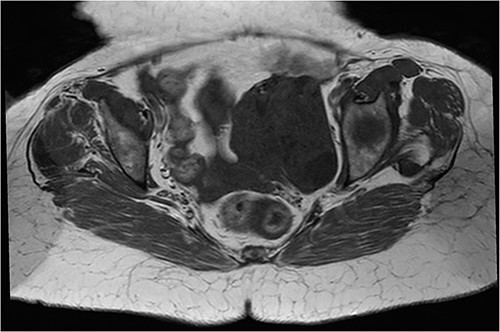
Axial T1 sequence: bulky left uterine mass with T1 hyposignal.
Biologically, the CA 125 tumor marker was at 126.2 IU/ml. The patient underwent a left adnexectomy and an omentectomy (small omentum). Macroscopically, the ovarian mass was smooth on the surface and fleshy on section with the presence of necrotic and hemorrhagic changes. On the histological level, the tumoral proliferation presents a diffuse architecture formed by sheets and clusters separated by fine fibrous septa. Tumor cells are rounded, small, and monomorphic with hyperchromatic, finely nucleolated nuclei and reduced cytoplasm. Mitotic activity is high (36 mitoses/10 HPF) (Figs 3 and 4). The samples taken from the small omentum were tumorous. An immunohistochemical study was carried out and showed diffuse positive expression by tumor cells of Vimentin, and focal by antibodies (Cytokeratin AE1/AE3, EMA, CD99, WT1, Inhibin, and SALL4) and an absence of expression by antibodies (CD45, Chromogranin, PAX8, and AFP) (Figs 5–7).
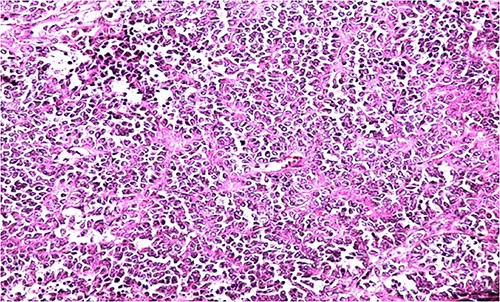
HE section showing diffuse sheets of small, closely packed round cells with scant cytoplasm separated by fibrous septa (x200).
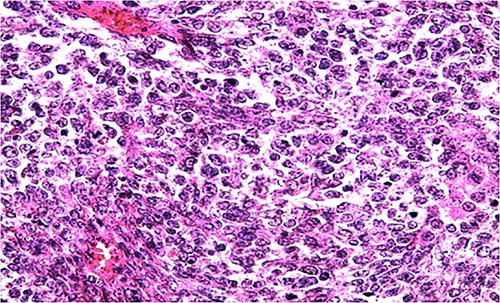
HE section of small monomorphic tumor cells showing hyperchromasia and mitotic figures (x400).
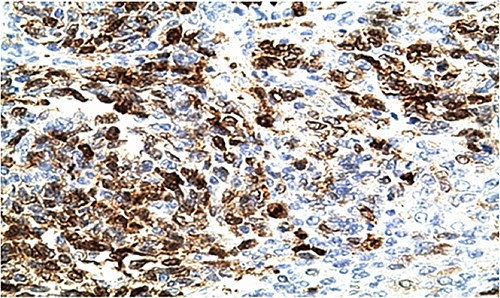
IHC: tumor cells show a nuclear strong expression of p16 (x400).
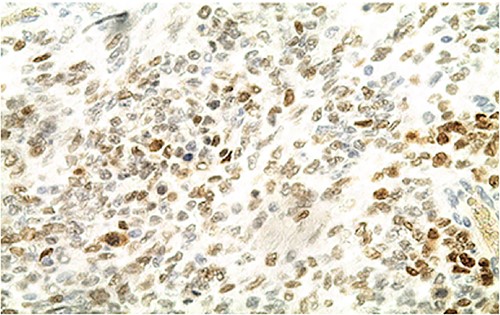
IHC: patchy nuclear expression of SALL4 by tumor cells (x400).
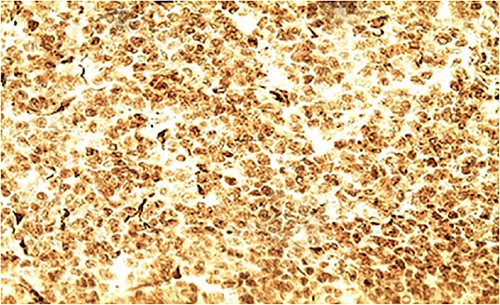
IHC: diffuse nuclear staining of WT1 by tumor cells (x400).
These immunohistochemical data ruled out lymphoma and do not argue in favor of a primitive neuroectodermal tumor or a juvenile granulosa tumor or a germ cell tumor.
The age, the clinical-biological context, and the anatomo-pathological data made it possible to conclude a diagnosis of ovarian small cell carcinoma of the hypercalcemic type, subsequently confirmed by a high dosage of Parathormone (4 ng/L).
Discussion
CPCOTH is extremely rare and usually affects young women. Since its discovery by Scully in 1979 [1], ~400 cases have been published in the literature [2].
The symptomatology is not specific, it is similar to other ovarian tumors. They are most often revealed by abdominopelvic masses or heaviness. About 66% of patients have hypercalcemia at diagnosis and 2.5% of them have clinical signs of hypercalcemia [3].
Grossly, these tumors are solid with a fleshy white pale to gray appearance, they are commonly large and unilateral, and we usually find areas of hemorrhage and necrosis [2].
The microscopic features consist of diffuse growth pattern made up of densely packed small cells and focal figures of follicle-like gaps with eosinophilic and, rarely, basophilic fluids. The majority of the cancerous cells are small and hyperchromatic. The nuclei have a single tiny nucleoli and chromatin that is loosely clumped. The mitotic activity is high. If the tumors are widely sampled, larger cells might be seen in at least half of all SCCOHTs. We name it “large cell variant of SCCOHT” when it is exclusively formed by large neoplastic cells. These cells have “rhabdoid” characteristics like eccentric nuclei with conspicuous nucleoli, and glassy eosinophilic cytoplasm. Small foci of mucinous glands or cysts walled by mature epithelium, signet-ring cells, or atypical mucinous cells are present in about 12% of cases [2].
In immunohistochemistry, the neoplastic cells occasionally show focal positivity for EMA, cytokeratins, calretinin, and CD10, whereas desmin, S100 protein, and inhibin are negative. Neuroendocrine markers are sometimes focally positive. Most cases are diffusely positive with WT1 antibody, which may be helpful for diagnosis even if many other tumors, including several in the differential diagnosis of SCCOHT, are also positive. Most of SCCOTH does not express hormone receptor-specific antibodies [4].
Multiple groups first characterized the genetic and molecular origins of SCCOHT in four seminal studies [5–8]. These articles revealed the critical function of SMARCA4 changes and demonstrated the pathogenic mutations in SMARCA4 that result in its biallelic inactivation as the primary cause of >90% of SCCOHT cases [5–7]. SMARCA4 has both somatic and germline mutations, with germline variants showing up in anywhere between 8% and almost 40% of instances, depending on the cohort [5–7]. Later, a thorough genomic analysis of SCCOHT demonstrated that these tumors had an astonishingly low number of somatic mutations and genomic stability, bolstering the idea that epigenetic dysregulation is primarily responsible for these tumors’ occurrence [9].
Although there is no universal agreement on the best course of action for SCCOHT, it typically entails multimodal chemotherapy, extensive surgery, and perhaps radiotherapy. The evidence that is now available, however, is made up of case reports or brief retrospective series with various management approaches, because no randomized studies have been done so far. A multicenter phase II trial at Institut Gustave Roussy is the only prospective clinical study in SCCOHT that tested a combination chemotherapy (PAVEP: cisplatin, adriamycin, vepeside, and cyclophosphamide) followed by radical surgery, high dose chemotherapy, and autologous stem cell transplant, and found that among the 27 SCCOHT patients, the 3-year survival rate was 49% [9].
Conclusion
SCCOTH is an infrequent neoplasm. Its rarity creates challenges in the diagnosis and management of affected patients. As pathologists, we must use molecular tools to sign the diagnosis. Surgery, chemotherapy, and radiotherapy are the basis of its treatment. Unfortunately, the prognosis remains poor despite the noticeable improvement in the management of these tumors [3, 4].
Conflict of interest statement
The authors declare that they have no competing interests.
Funding
No external funding sources are relevant to this submission.
Consent for publication
Written informed consent for publication of their clinical details and/or clinical images was obtained from the patient.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}