





Abstract
Primary hepatic rhabdomyosarcoma is an exceedingly rare type of sarcomas. A 15-year-old boy was admitted to the hospital with abdominal pain. Serial investigation showed a giant heterogeneous mass, with a diameter of 15 cm, located in the right lobe of the liver (segment 4a, 4b, 5, 6, 7, 8), with clear margins, non-homogeneous density in computerized tomography and the Positron Emission Tomography Scan Multi Slice revealed a peripherally slightly metabolically active hepatic mass (205 × 134 × 208 mm). Histopathological examination and the Immunohistochemical showed embryonal rhabdomyosarcoma. The management strategy involved multiple cycles of ifosfamide and doxorubicin and monitoring the tumor size, until the tumor size was suitable for the surgery. The management of hepatic rhabdomyosarcoma is a surgical challenge due to the late onset of symptoms, a limited number of reported cases, and poor prognosis. Surgical excision is the treatment of choice for primary hepatic rhabdomyosarcoma and combination between chemotherapy, surgery showed good results.
Introduction
Sarcomas are rare group of malignant tumors that originate from mesenchyme, and have >70 pathological types [1].
Rhabdomyosarcomas make up the largest type of soft-tissue sarcomas in children and adolescents, representing a proportion of 5 to 10% of all solid tumors in childhood, and it is rare among adults [2].
Rhabdomyosarcomas mainly have four tissue types: embryonal, alveolar, pleomorphic, and spindle cell, and it can affect any organ, mainly affects head and neck, genitourinary organs, retroperitoneum, and extremities [2–4].
Primary hepatic rhabdomyosarcoma is exceedingly rare, and ~20 cases have been reported in the literature since 1956 [5, 6], and it is known that rare types of sarcoma are associated with high mortality percentages due to delaying in diagnosis and management [7].
Here we present an interesting case of clinical management and follow-up of primitive hepatic embryonal rhabdomyosarcoma in a 15-year-old male.
Case presentation
A 15-year-old boy was admitted to the hospital by his parents with abdominal pain for the past month. The pain was located in the right upper quadrant and radiated to the right shoulder and lower back. This patient had no significant family, surgical, or medical history. Laboratory data were normal, except for an elevated Alkaline Phosphatase and Urea.
Abdominal and pelvic ultrasonography revealed an enlarged liver with a diameter of (~15 cm). Most of the right lobe was occupied by a round, heterogeneous mass measuring (153 × 156 × 160 mm), which contained areas of necrosis and a fluid-filled cavity. On Doppler ultrasound, clear arterial vascularity was observed within the mass.
A computerized tomography (CT) scan of the chest, abdomen, and pelvis with intravenous contrast medium confirmed the presence of this giant heterogeneous mass, with a diameter of 15 cm, located in the right lobe of the liver (segment 4a, 4b, 5, 6, 7, 8), with clear margins, non-homogeneous density, that does not contain obvious calcifications. It showed peripheral enhancement with delayed central nodular enhancement without accompanying cysts. No dilation was observed in the biliary or portal ducts. There was a mild reactive pleural effusion with no evidence of secondary pulmonary or hepatic lesions (Fig. 1).
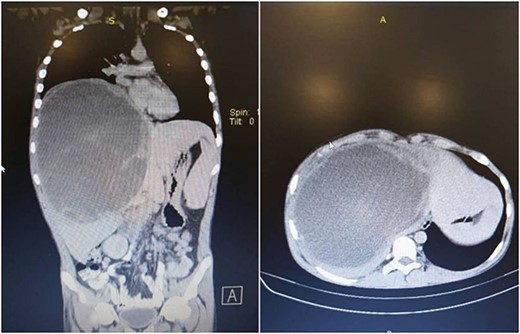
Chest, abdomen, and pelvis CT scan showed a giant heterogeneous mass in the right lobe of the liver.
In succession, the Positron Emission Tomography (PET) Scan Multi Slice revealed a peripherally slightly metabolically active hepatic mass (205 × 134 × 208 mm) with a wide necrotic component and inactive mesenteric, retroperitoneal, and pulmonary nodes (Fig. 2).
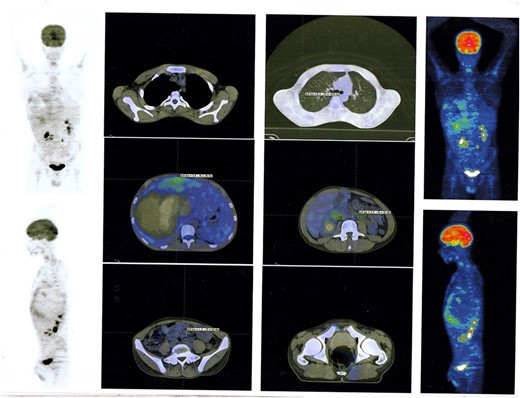
PET Scan Multi Slice revealed a peripherally slightly metabolically active hepatic mass (205 × 134 × 208 mm) with a wide necrotic component and inactive mesenteric, retroperitoneal, and pulmonary nodes.
The patient underwent an ultrasound-guided fine needle aspiration. Histopathological examination of the tumor cells displayed pleomorphic cells with a high n/v ratio consistent with mesenchymal origin. Immunohistochemical expression showed positivity for desmin and vimentin and negativity for hep par and CD 138 (Fig. 3).
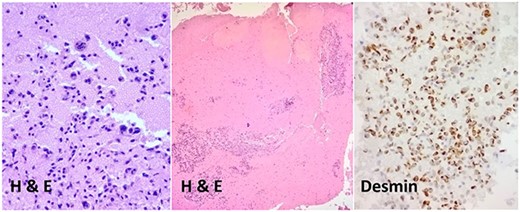
Histopathological examination of the tumor cells.
The diagnosis was embryonal rhabdomyosarcoma based on the immunohistochemical result.
Following a multidisciplinary consultation, it was determined that surgical excision of the tumor was not feasible.
Chemotherapy consultation advised three cycles of ifosfamide and doxorubicin (each cycle for 21 days). Then, multislice computerized tomography (MSCT) was requested that revealed the transformation of the solid hepatic lesion into a cystic lesion with necrotic regions, with minor increase in the lesion size. Prolonged the ongoing chemotherapy treatment for five additional cycles was recommended due to the high surgical risk, furthermore, the size of the tumor is very large and the remaining portion of the left lobe is deemed inadequate for sustaining life.
New MSCT showed a significant improvement in lesion size, with a 10% reduction in tumor size.
After surgical consultation, we decided to extend the chemotherapy treatment for two additional cycles before surgical excision because the size of the tumor was still large despite the improvement.
Right extended hepatectomy (the 4a, 4b, 5, 6, 7, 8 segments) with cholecystectomy were performed. The hepatic lobe weighed 1700 g and measured 190 × 170 × 150 mm (Fig. 4). Sections showed somewhat well-defined total necrotic mass measuring ~14 cm in the largest dimension, not reaching the hepatic capsule surface. Multiple sections were submitted for histologic examination, lymph node measuring 1.5 cm. The entire specimen was embedded for histologic examination, the gallbladder measured 7 × 4 cm and showed 0.7 cm black stone, and no masses were seen.
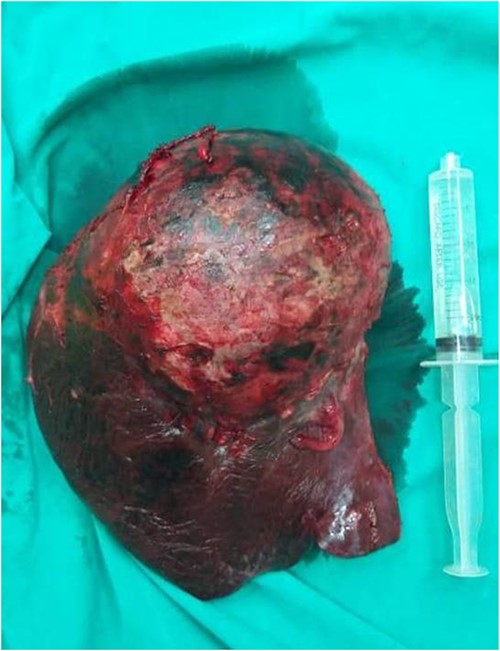
A gross examination of the right hepatic lobe specimen.
The histologic examination of the excised lesion revealed an entirely necrotic liver tissue mass due to a total burnt-out tumor with excellent treatment response. No viable tumor cells were seen. The remaining hepatic tissue showed no significant pathologic changes, and all surgical margins were free of neoplastic changes. The hepatic hilar lymph node was free of tumor (0/1) and mild chronic cholecystitis and cholelithiasis were seen in the gallbladder.
The patient was followed up every 3 months with serial MSCT.
One month after surgery, an abdomen MRI revealed a fluid-filled cyst measuring (1.7 × 3 cm) located behind the liver, between the inferior vena cava and the caudate lobe of the liver without enhancement after injection (most likely postoperative complications). We decided to give the patient three additional cycles of the same chemotherapy strategy. After completing the three additional cycles of chemotherapy, we performed MSCT scan with contrast injection that revealed a reduction in the size of the hepatic fluid-filled cyst without metastases.
The overall treatment time that includes surgical treatment and chemotherapy was nearly 11 months. After following up with the patient for one year, PET Scan Multi Slice showed normal findings without signs of local recurrence, without nodal or visceral metabolic activity (Fig. 5). Regular clinic visits and periodic check-up every 6 months for 3 years showed the patient in good health without signs of recurrence.
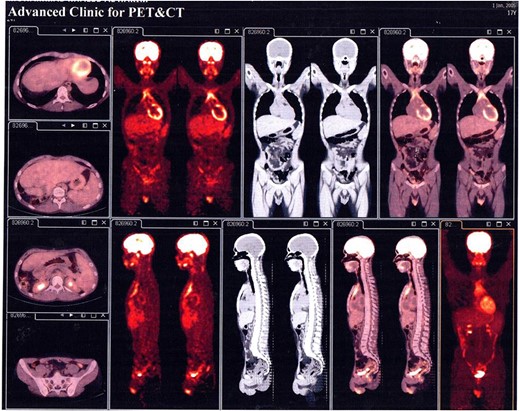
PET Scan Multi Slice showed normal findings without signs of local recurrence, without nodal or visceral metabolic activity.
Discussion
Rhabdomyosarcomas mainly have four tissue types: embryonal, alveolar, pleomorphic, and spindle cell, and it can affect any organ. The most common types are embryonal and alveolar and they usually affect adolescents, whereas pleomorphic occurs in adults, and spindle cell can be found in adults and children [2].
Hepatic primary rhabdomyosarcoma is exceedingly rare, and ~20 cases have been reported in the literature since 1956. The main manifestations of patient with hepatic primary rhabdomyosarcoma are epigastric pain, fever, abdominal distension, anorexia, vomiting, fatigue, and shortness of breath, and the patient may be asymptomatic until the tumor size becomes larger [6, 8].
Our patient presented with abdominal pain located in the right upper quadrant and spread toward the right shoulder and lower back.
The diagnosis is made using biochemical, radiologic, and histological studies.
Biochemical studies may show elevated alpha-fetoprotein or signs of cholestasis, but in general, it is not significant in the final diagnosis. In addition, radiographic studies have shown great benefit in a few numbers of cases, and knowledge of characteristics MRI of mesenchymal liver tumors played an important role in reducing the range of potential diagnoses for the pathologist. The histological studies are considered as the gold standard for the diagnosis [8].
Histological studies of embryonal rhabdomyosarcoma revealed primitive mesenchymal cells in various stages of myogenesis in eosinophilic cytoplasm. Immunohistochemical analysis plays a crucial role in distinguishing the different types.
According to Li et al., the vimentin marker was positive in most cases, and nuclear staining using MyoD1 and myogenin has high sensitivity and specificity for rhabdomyosarcoma samples [9].
Our sample was positive for vimentin marker during histopathological studies.
The management of hepatic rhabdomyosarcoma is a surgical challenge due to the late onset of symptoms, a limited number of reported cases, and poor prognosis.
Li et al. found that patients who went through surgery had a better survival time of 16.7 months, compared with patients with non-surgical therapy had only 9.5 months, in addition, patients with larger tumors had shorter survival times [9].
The surgical technique included anatomic lobectomies (left/right hemihepatectomy or extended hemihepatectomy) [10].
He also explained the importance of the combination of neoadjuvant chemotherapy, surgery, prophylactic radiotherapy, and polychemotherapy. In general, chemotherapy and radiotherapy alone showed low sensitivity in adult patients’ therapy [10].
Our patient underwent a complex management strategy to reduce the tumor size, which started with several cycles of chemotherapy followed by monitoring with MSCT scans to assess tumor reduction, then surgical excision.
The age considered as prognostic factor for rhabdomyosarcoma patients. They have poor prognoses in general. The survival rate of adult patients with rhabdomyosarcoma is 20–40% in comparison with the children group is 70%, In addition, children under 1 year and over 10 have a worse prognosis [10, 11].
Conclusion
Hepatic primary rhabdomyosarcoma is exceedingly rare with a poor prognosis. In our case, chemotherapy, especially for large tumors, followed by surgery, showed good results on survival and improving the patient’s condition.
Conflict of interest statement
None declared.
Funding
None declared.
Data availability
All data are included in this published article and its supplementary information files.
Author contributions
M.Z.B.A., R.G.: analyzed and interpreted the patient data, wrote the manuscript, and revision.
M.A.N.: the vascular surgeon, revision, and patient care.
E.M.C.: The pathologist, critical review, and patient care.
H.A.H., Z.N.O.: The oncologist, revision.
F.H.: The general surgeon, supervision, and patient care.
All authors read and approved the final manuscript.
Consent for publication
Written informed consent was obtained from the patient’s legal guardian for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}