





Abstract
Retroperitoneal ganglioneuroma (RGN) is a rare benign tumor that arises from the retroperitoneal sympathetic ganglia composed of mature Schwann cells, ganglion cells and nerve fibers. These tumors can occur anywhere along the paravertebral sympathetic plexus and occasionally from adrenal medulla. Although they grow in excessive size, they may cause compression to adjacent organ or structures thus giving rise to symptoms. Resecting RGN’s is a challenging endeavor, as they tend to encase neighboring vessels to their site of origin. The reported case is a 43-year-old male who presented with lumbar pain that increased progressively in intensity over the last 6 months. Preoperative investigations revealed a large tumor with encasement of the origins of the superior mesenteric artery and bilateral renal arteries. The tumor was completely resected and the final pathology confirmed the diagnosis of RGN.
INTRODUCTION
Retroperitoneal ganglioneuromas (RGN’s) are rare benign tumors originating from the retroperitoneal sympathetic ganglia. They are considered neuroblastic tumors along with ganglioneuroblastomas and neuroblastomas, with the differential being based upon the degree of cellular and extracellular maturation [1]. The definitive diagnosis can be achieved on the basis of pathological examination from a biopsy or the resected specimen [1]. They are usually sporadic; though an association with neurofibromatosis types II and multiple endocrine neoplasia (MEN) IIB has been reported [2]. RGN’s are usually diagnosed in the age group of 20–35 years, with a similar incidence between sexes. RGN’s are typically asymptomatic until they grow to significant size where they tend to cause symptoms secondary to compression of adjacent organs or vessels, thus making complete tumor resection challenging. We present a case of a gigantic retroperitoneal tumor in a male patient who presented with lower back pain, which was confirmed to be a RGN in the final pathology report.
CASE REPORT
A 43-year-old Caucasian male with an unremarkable past medical history was referred for a retroperitoneal tumor diagnosed on a computerized tomography (CT) scan performed for progressively worsening lower back pain. Physical examination was unremarkable and the laboratory values including tumor markers were within normal range. The abdominal CT scan demonstrated a large solid non-enhancing retroperitoneal mass, with a well-defined margin, measuring 9.6 cm × 9.3 cm, located anteriorly to the aorta and displacing the inferior vena cava (IVC), pancreas, the superior mesenteric artery (SMA) and the renal vessels (Fig. 1). Patient underwent an exploratory laparotomy and a tumor was found lying in the retroperitoneum adherent to aorta, IVC, SMA, pancreas and renal vessels (Fig. 2). Complete right medial visceral rotation was performed and the small bowel mesentery was mobilized exposing the retroperitoneum. Meticulous dissection of the tumor from the aorta IVC and their main abdominal branches was performed and the specimen was excised. The specimen consisted of a well-circumscribed, solid tan to yellow tumor measuring ~9 × 9 cm encased by a fibrous capsule. Histological, no atypia or mitotic activity was evident and the tumor was composed of variably sized mature ganglion cells with satellite cells deposited in a neuromatous stroma. All three lymph nodes included within the surgical specimen were not infiltrated. The ganglion cells stained positive for S-100 protein, glial fibrillary acidic protein and neuron-specific enolase (NSE) while Ki-67 labeling index was <1% (Fig. 3). The final pathology report was RGN. Postoperative course was uncomplicated. Patient is well on a 2-years follow-up without clinical or radiographic evidence of recurrent disease.
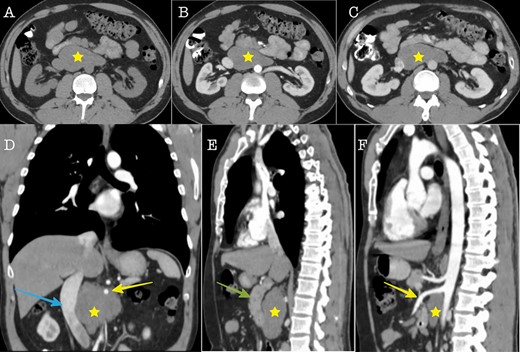
Cross sectional imaging (A): Axial CT without contrast medium, (B) Axial CT contrast-enhanced, arterial phase, (C) Axial CT, contrast-enhanced, portal venous phase. The images show a large solid retroperitoneal mass (yellow star) isoattenuating to the muscles, without contrast enhancement. (D) Coronal reconstruction and (E, F): Sagittal reconstructions demonstrate the anatomic relationships of the mass with IVC (blue arrow), superior mesenteric artery (yellow arrow) and pancreas (green arrow). The mass is located dorsally to the pancreas, encases the superior mesenteric artery and displaces the IVC to the right.
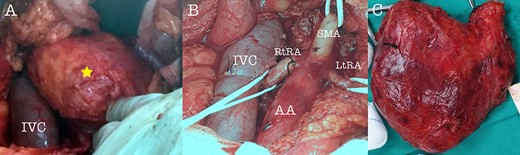
Intraoperative view of the tumor (A), after tumor resection (B) and tumor specimen (C), (AA: abdominal aorta, RtRA: right renal artery, LtRA: left renal artery, IVC: inferior vena cava).
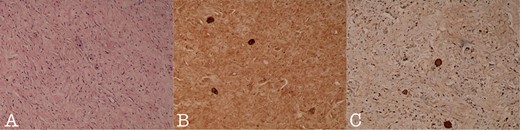
Histology section of (A): Ganglioneuroma composed of variably sized mature ganglion cells with satellite cells deposited in a neuromatous stroma (H-Ex100), (B): NSE staining of the ganglion cells (NSEx100) and (C): S100p staining of the gaglion cells and the schwannian stroma (S100px100).
DISCUSSION
Ganglioneuromas are extremely rare benign tumors originating from the neural crest tissue, which gives rise to the sympathetic ganglia. These tumors can arise from sympathetic nervous tissue at any anatomical site; the most common site being the retroperitoneum (32–52%), posterior mediastinum (39–43%) and neck (8–9%) adjacent to the spinal cord. Among abdominal ganglioneuromas, 49% originate within the adrenal gland and 51% are extra-adrenal. Their incidence in the general population is about one per million and they are usually sporadic [3].
RGNs are usually asymptomatic and as they increase in size they compresses and displaces adjacent tissues, organs and/or vessels, thus causing symptoms, which are site specific. Common symptoms include abdominal pain, abdominal distention, fatigue or lower-back pain, as in our case. Rarely, they may exhibit secretion of vasoactive intestinal polypeptides and/or catecholamine, giving rise to symptoms such as diarrhea, sweating and hypertension [4]. Due to lack of specific characteristics, RGNs are quite difficult to differentiate from other tumors including neurofibroma, schwannoma, neuroblastoma, ganglioneuroblastoma and pheochromocytoma [5].
Imaging features are of particular importance in assessing RGN’s. Ultrasound examination indicates usually a homogenous, well-defined, hypoechoic mass, whereas contrast-enhanced CT reveals a homogenous relatively well-encapsulated mass with no or slight enhancement in arterial phase and progressive mild enhancement in delayed phase. In 20% of patients circumscribed or spotted calcifications may be seen. On magnetic resonance imaging the tumor is described as isointense or hypointense with the spinal cord on T1-weighted images, hyperintense on T2-weighted images and also as heterogeneous on contrast-enhanced T1-weighted images. RGNs have no fluorodeoxyglucose uptake thus rendering positron emission tomography and computed tomography of no diagnostic value [3]. Although it is not feasible to categorically differentiate between benign and malignant neural crest tumors, imaging findings provide valuable information about the tumor extent, its regional invasion, organ origin, calcification and adenopathy [4].
Fine Needle Aspiration (FNA) may be used preoperatively in selected cases; however it may be misleading in terms of diagnosis. Since RGN’s rarely secrete catecholamines, a Fine Needle Aspiration can be performed with relative impunity from a catecholaminergic crisis [5]. In histopathological examination, typical characteristics of RGNs are nerve fibers and mature sympathetic ganglion cells [6]. Immunohistochemical reactions are usually positive for S-100 protein and negative for desmin, smooth-muscle actin and CD34 [6].
Since GN’s are not associated with ganglioneuroblastoma changes and seem to have benign progression, perioperative radiotherapy or chemotherapy are not useful if complete surgical resection is achieved (6). Tao et al. found that an AKT–mTOR–S6 pathway was active in human ganglioneuroma. The authors proposed the potential clinical impact of mTOR-targeting drugs on clinical management of ganglioneuroma patients, particularly those that are deemed surgically unresectable [7].
The treatment of choice for GNs is complete surgical resection [8–10]. Preoperative distinction of ganglioneuromas from other aggressive tumors is prudent since their non-infiltrative nature will allow for organ/vessel sparing surgery, thus avoiding unnecessary morbidity from an over ambitious surgery. Complete resection however remains the most robust prognostic factor for disease free survival [11, 12]. Nevertheless, given the indolent course of GNs with slow and quiescent progression, regular and long-term follow-up is recommended to monitor for local recurrence.
CONCLUSIONS
Retroperitoneal GNs are rare well-differentiated slow growing tumors, usually presented without any significant symptoms. Although they can grow extremely large, rarely invade and more often displace adjacent vessels and organs. Complete surgical resection is the treatment of choice although technically challenging given its anatomic origin. Recurrence is rare after complete resection. Regular and long-term follow-up is recommended to exclude local recurrence especially in cases with microscopic residual disease.
CONFLICT OF INTEREST STATEMENT
None declared.
FUNDING
None.

{kind=link}
{kind=link}
{kind=link}