





Abstract
Efficient extraction of knowledge from biological data requires the development of structured vocabularies to unambiguously define biological terms. This paper proposes descriptions and definitions to disambiguate the term ‘single-exon gene’. Eukaryotic Single-Exon Genes (SEGs) have been defined as genes that do not have introns in their protein coding sequences. They have been studied not only to determine their origin and evolution but also because their expression has been linked to several types of human cancer and neurological/developmental disorders and many exhibit tissue-specific transcription. Unfortunately, the term ‘SEGs’ is rife with ambiguity, leading to biological misinterpretations. In the classic definition, no distinction is made between SEGs that harbor introns in their untranslated regions (UTRs) versus those without. This distinction is important to make because the presence of introns in UTRs affects transcriptional regulation and post-transcriptional processing of the mRNA. In addition, recent whole-transcriptome shotgun sequencing has led to the discovery of many examples of single-exon mRNAs that arise from alternative splicing of multi-exon genes, these single-exon isoforms are being confused with SEGs despite their clearly different origin. The increasing expansion of RNA-seq datasets makes it imperative to distinguish the different SEG types before annotation errors become indelibly propagated in biological databases. This paper develops a structured vocabulary for their disambiguation, allowing a major reassessment of their evolutionary trajectories, regulation, RNA processing and transport, and provides the opportunity to improve the detection of gene associations with disorders including cancers, neurological and developmental diseases.
Introduction
Next-Generation Sequencing (NGS) and other high-throughput technologies are generating vast amount of biological data that are a challenge for downstream data mining. To help address this problem, progress has been made in the development of structured vocabularies and ontologies that facilitate computational data annotation, retrieval and interpretation (1–4). However, no such structured vocabulary exists for describing the different types of eukaryotic single-exon coding sequences (CDSs), causing confusion and leading to misinterpretation of their evolutionary origins as well as their regulation and function within eukaryotic genomes.
This situation is being further exacerbated by the discovery that Single-Exon Isoforms (SEIs) are being misannotated as arising from Single-Exon Genes (SEGs) rather than from Multi-Exon Genes (MEGs) by alternative splicing as is the case. It is urgent to draw the attention of the scientific community to such problems before such errors become indelibly propagated in biological databases. ‘This work attempts’ to address these concerns by proposing a workflow for developing a structured vocabulary (see also the glossary box) and ontology to describe and distinguish the various types of eukaryotic single-exon CDSs; one that is resilient and inclusive but flexible enough to accommodate new advances in gene interpretation. The workflow is presented as a directed acyclic graph with transitive rules for deriving ontological descriptors (Figure 1).
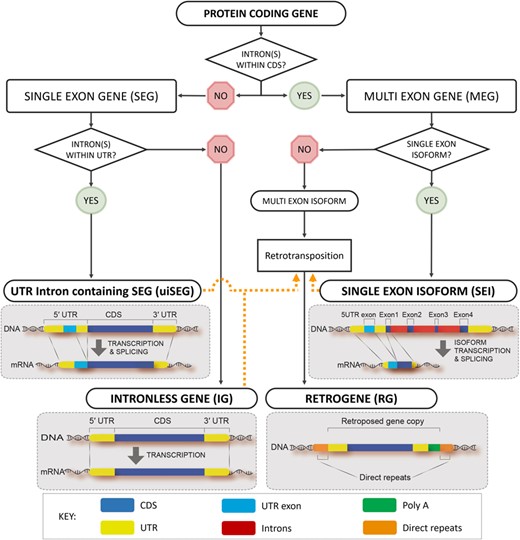
Workflow for developing a structured vocabulary (ontology) to distinguish different types of single exon CDSs. UTR intron containing genes (uiSEG); Intronless Gene (IG); Single Exon Isoform (SEI) and Retrogene (RG). Orange dotted lines connect genes to retrogenes via retrotransposition processes. CDS = protein coding region; UTR = untranslated region. The orange dotted lines connect RGs with potential parental genes. RGs are synonymous with IGs only when the retrotransposition origin of these sequences can be implied.
Single-exon genes
Eukaryotic genes are usually interrupted by intragenic, non-protein coding regions termed introns that are removed by RNA splicing during maturation of the final RNA product. However, more than 2000 protein-coding genes in human genome have been shown to lack introns and have been termed SEGs, defined as a nuclear, protein-coding gene that lack introns in their CDSs (5). This definition excludes genes that generate functional RNAs such as tRNA, rRNA and long non-coding RNAs. A large proportion of genes encoding G-protein-coupled receptors (GPCRs), especially the olfactory receptors, the major subfamily of class A GPCRs (6), and genes encoding canonical histones (7) are known to be SEGs. It has been proposed that the expression of many human SEGs is linked to several types of cancer and neurological and developmental disorders (8). In addition, the expression of some SEGs is testis and neuro-specific (8, 9). These discoveries highlight the importance of studying SEGs to uncover properties and evolutionary trajectories that underlie their relationships with both pathologies and normal phenotypes.
As shown in Figure 1 (left side), SEGs can be divided into the following two main groups: (i) SEGs having introns in their untranslated region (UTR), so-called “UTR intron-containing SEGs” (uiSEGs) and (ii) SEGs lacking introns in the entire gene, termed ‘intronless genes’ (IGs) (10) (see also the glossary box for definitions).
Examples of uiSEGs with experimentally validatedphenotypes of clinical relevance are as follows: ERAS, embryonic stem cell expressed Ras; NEUROD2, neuronal differentiation 2; and NFIL3, nuclear factor, interleukin-3-regulated protein (5, 11, 12).
Examples of SEGs that can be classified as IGs are as follows: Reprimo (RPRM), a TP53 dependent G2 arrest mediator (10, 12, 13); CDR1, cerebellar degeneration-related protein; and NPBWR2, neuropeptides B/W receptor type 2 (10, 11).
Ontological ambiguity
There is a significant operational problem in distinguishing between uiSEGs and IGs. Today, most genes are predicted based mainly on bioinformatics analyses. Inparticular, SEGs are identified based on CDS (protein coding) gene identifiers in annotated genomes (5, 14–16) and these identifiers do not include information from the UTR of genes, resulting in the identification of all SEGs as IGs, regardless of the presence or absence of introns in their UTRs (14–18) with the result that the terms ‘SEG’ and ‘IG’ are rife with ambiguity.
The distinction between uiSEGs versus IGs is important to make because the presence or absence of introns in the UTR, and whether the intron is in the 5′ UTR or 3′ UTR, can impact transcriptional regulation and post-transcriptional processing (19–21). For example, RNA transcripts derived from 5′ UTR intron-containing genes (35% of all human transcripts) (22) are exported from the nucleus by a splicing-dependent mechanism involving the TRanscription and EXport (TREX) complex (20). TREX is a conserved multi-subunit complex that is recruited to the 5′ end of mRNA transcripts by capping and splicing events (23). The TREX export pathway has been implicated in several diseases (23). On the other hand, RNA transcripts lacking 5′ UTR introns, such as IGs, can harbor specific sequences in their early coding regions (24) that promote an alternative mRNA nuclear export pathway (20). The majority of these mRNAs encode secreted, membrane-bound or mitochondrial proteins (25). The presence or absence of introns in the 5′ UTR of genes has also been shown to affect transcriptional activity, protein accumulation and determination of tissue-specific transcription (26, 27), providing another example of the need for disambiguation of uiSEGs from IGs.
Less is known about the function of 3′ UTR intron-containing genes, although some have been shown to target mRNA for degradation by the nonsense-mediated decay pathway (19, 27). It has also been observed that 3′ UTR introns can modulate gene expression at multiple levels and has been associated with miRNA targets (28) and specific mRNA localization in neurons (29, 30).
These data together suggest that in order to clearly distinguish between uiSEGs and IGs, an experimental validation of UTR introns should be considered.
SEGs are not always synonymous with retrogenes
Retrogenes (RGs) (Figure 1, bottom right-hand side) arise by retrotranscription of mRNA followed by insertion of the resulting DNA copy into the genome (31, 32). Most RGs are believed to have originated from multi-exon (intron-containing) parental genes (33) (Figure 1) although theoretically they could also arise from mRNA derived from uiSEGs and IG transcripts (Figure 1, dotted arrows). RGs are generally thought to be intronless but recently, intron-containing RGs derived from retrotransposition of parental isoforms with retained introns (31, 34) and MEGs including RG-derived exons (31) has been discovered.
Although many SEGs are thought to be RGs (14), molecular mechanisms distinct to retrotransposition have been proposed for the origin of SEGs, such as de novo origin (35), DNA-based duplication from intron-containing genes (36) and intron loss, among others (37, 38). Clusters of genes encoding canonical replication-dependent histones that evolved to possess a specialized 3′ processing pathway that is coupled to DNA replication (39, 40) are remarkable examples of SEGs that are not RGs (31, 41).
Initially, RGs may exhibit sequence signatures of their mRNA origin and genome insertion such as poly-A tails and direct repeats. But these molecular signals may become blurred over time that could impact the annotation of RGs. Furthermore, many RGs contain mutations that may render them inactive and are termed ‘processed pseudogenes’ (31, 42).
These data together suggest that despite their similar molecular structure, RGs are synonymous with SEGs only when the retrotransposition origin of the sequence can be implied.
Single-exon isoforms
A previously undefined class of mRNA transcripts, which we term SEIs, is similar to mRNAs derived from SEGs (9). However, unlike mRNA from SEGs, SEIs originate by alternate splicing of RNA transcribed from MEGs (Figure 1, top right-hand side and Supplementary Figure S1) in which only one protein coding exon is retained in the mature mRNA (SEI). The gene structure, transcriptional regulation and evolutionary origin of SEIs differ radically from SEGs.
The emerging problem is that SEGs and SEIs are being confused because bioinformatics techniques for gene identification based on CDS annotation do not take into account the underlying gene structure. To aid in the resolution of this problem, we suggest that a closer examination of the underlying gene structure at the DNA level would facilitate the distinction between a single-exon transcript that arose from a SEG (uiSEG or IG) and one that arose from a MEG by alternate splicing (SEI).
Analysis of the human genome (GRCh38.p9 Refseq assembly GCF_000001405.35) using a genomics protocol described in Supplementary Material, predicted 2783 putative SEGs of which 621 (22.3%) correspond to predicted SEIs and 38 sequences (1.4%) were identified as potential annotation errors. Using manually curated methods, 687 sequences have been previously predicted to be IGs (10), these data together let us to estimate that an approximate of 1437 (51.6%) predicted SEGs—using our protocol—correspond to uiSEGs (Supplementary Figure S2).
The estimated proportion of uiSEGs, IGs and SEIs within the human genome is about 2:1:1 and the latter group could have caused bias in earlier studies of SEG function and evolution in the human genome (5, 8, 14–17, 43). The unambiguous identification of SEIs also provides opportunities to study novel aspects of alternative splicing and intron evolution, gene evolution, RNA editing, nuclear export pathways and transcriptional regulation.
Examples of SEIs with experimental validation and clinical relevance that have previously been classified as SEGs include the following: BDNF, brain-derived neurotrophic factor isoform a preproprotein; HIC1, hypermethylated in cancer 1 protein isoform 1; and GDNF, glial cell line-derived neurotrophic factor isoform (11, 12).
Implementation
The proposed structured vocabulary has been submitted to a publicly accessible ontology project: Sequence Ontology (SO, http://www.sequenceontology.org/).
Conclusions
Eukaryotic SEGs can be divided into the following two main groups: IGs and uiSEGs. This distinction is important to make because the presence or absence of introns in the UTR, can impact transcriptional regulation and post-transcriptional processing of the mRNA. In order to understand their evolution and biology, it is important to clearly distinguish between the different types of genes.
Despite similar molecular architecture between some RGs and SEGs, these terms are synonymous only when the retrotransposition origin of the sequence can be implied.
A previously undefined class of mRNA transcripts, which we term SEIs, is being incorrectly annotated as SEGs, exacerbating the operational problem in distinguishing the different types of SEGs.
With the increase of RNA sequencing approaches, this confusion is likely to become aggravated if it is not solved. This task is urgent so as to reduce the propagation of erroneous gene annotations in biological databases.
A structured vocabulary with unambiguous definitions of SEGs, IGs and SEIs provides opportunities to study novel aspects of alternative splicing and intron evolution, gene evolution, RNA editing, nuclear export pathways and transcriptional regulation. It also provides the opportunity to improve the detection of gene associations with disorders including cancers, neurological and developmental diseases.
Funding
Fondecyt (1090451, 1130683, 1181717) and Basal (AFB170004).
Conflict of interest. None declared.
Database URL:http://www.sinex.cl
References

{kind=link}